|
1. Corresponding author, e-mail: boya@chemeng.Lan.mcgill.ca Abstract
1. Introduction Uranium is one of the most seriously threatening heavy metals mainly because of its high toxicity, not so much radioactivity. Uranium contamination poses a threat in some surface and groundwaters [1, 2]. Activities associated with the nuclear industry have brought excessive amounts of uranium into the environment [3]. Biosorption has been proposed as a potential alternative for removing toxic heavy metals. Various biosorbents based on non-living biomass have demonstrated an excellent uranium adsorption performance. For example, filamentous fungi, yeast, bacteria, actinomycetes and fresh-water algae, such as Chlorella, have been reported binding uranium in excess of 150 mg/g of dry biomass [4, 5, 6, 7, 8, 9, 10]. Marine algae represent an important biosorbent type. They proliferate ubiquitously and abundantly in the litoral zones of world oceans often posing environmental nuisance. Metal biosorption capacities of non-living seaweed biomass were summarized by Kuyucak and Volesky [11]. The brown alga Sargassum fluitans has been is particularly effective in binding heavy metal ions of gold, cadmium, copper, zinc, etc. [12]. The high sorption capacity, easy regeneration and low-costs make this biomass of special interest for purification of large volumes of wastewater with lower concentration levels of metal toxicity to be removed [12, 13, 14, 15]. This is either difficult and/or expensive to accomplish by conventional metal-removal processes. Although the fact that marine algae are capable of biologically concentrating radionuclides such as radium, thorium and uranium has been known for a long time [16], the biosorption of uranium by non-living marine algae has not been reported. The present work extended investigations of metal biosorption by the protonated Sargassum alga biomass to the uranium removal and recovery. In this work, basic parameters of equilibrium uranium biosorption were determined and the biosorption-desorption in a flow-through packed column was examined. 2. Materials and Methods 2.1. Preparation of sorbent. Beach-dried Sargassum fluitans, collected in Naples, FL, was chopped up in a homogenizer and sieved to different fraction sizes. The batch of dry biomass with particle size (1.0 - 1.4) mm was selected for subsequent protonation pretreatment aimed at standardizing the biomass by eliminating the light metals Ca2+, Mg2+ etc. The protonation wash using 0.1 N HCl (10 g biomass / L ) resulted in some biomass weight loss. After 3 hours of contacting with acid, the biomass was rinsed with deionized water in the same volume many times until a stable wash solution pH 4.0 was reached. The biomass was then dried in an oven at 40-60 OC overnight. So prepared biomass was stored for later use.
The initial uranium concentrations Ci
correspond to the control samples, and the final uranium concentrations
Cf were from the supernatant solution. The uranium uptake
was calculated by the concentration difference method that is based on
the mass balance as follows:
with V being the solution volume, and W being the mass of biosorbent. In the desorption experiments, 0.1 g of metal-loaded biomass was mixed with 50 mL 0.1 N HCl in a 100 mL Erlenmeyer flask. The remaining procedure was the same as that in the sorption equilibrium experiments except that no pH adjustment was required. The eluted biomass metal content could be calculated directly from the amount of metals desorbed into the HCl solution as follows: qdes.= Cdes. V / W (2) with q des. being eluted metal content per gram of biomass, and C des. being the metal concentration in the HCl eluent solution.
3. Results
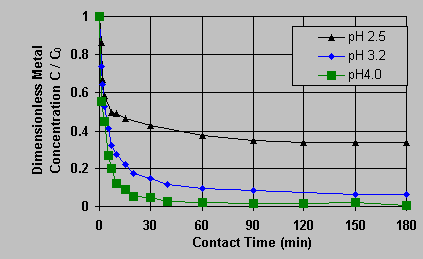 C0 = 190.5 mg/L. The uranium biosorption rate was strongly influenced by the sorption system pH value, the uranium solution concentration in the solution decreased with contact time faster at higher pH values. At various pH values, approximately 70 - 80% of the uranium present originally in the solution was sorbed onto the biomass in about 15 minutes after the start of biosorption and the equilibrium could be reached within 3 hours. This provided a guide for the biosorption contact time to be used in the following equilibrium experiments. The equilibrium of the uranium biosorption on Sargassum biomass was expressed in resulting biosorption isotherms for pH 2.6, pH 3.2 and pH 4.0 (Figure 2). 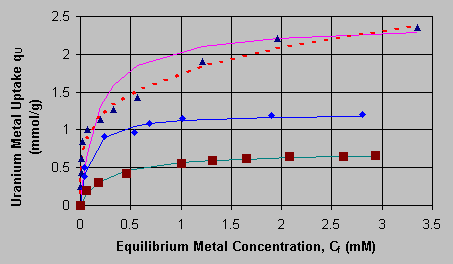 (s ) pH 2.6; (u ) pH 3.2; (n ) pH 4.0; () Langmuir model; (---) Freundlich model The lines in Figure 2 refer to model-calculated values and the points are for experimental uranium uptakes. At pH 2.5 and pH 3.2, the isotherms could be represented well by the Langmuir sorption model, q = qm*Cf /(K + Cf), while the Freundlich model, q = k*(Cf)^n, could represent a better regression than the Langmuir model at pH 4.0. The model parameters, the maximum uptake capacities qm and the equilibrium constants K in the Langmuir model as well as k and n in the Freundlich model, were regressed from the experimental data at various pH values and are listed in Table 1: Table 1: Regressed Langmuir and Freundlich
sorption isotherm model parameters
Table 1 shows that qm and K were largely dependent on the final solution pH values. The fact that qm increased while the K decreased for higher solution pH values indicated that the sorption affinity of uranium for the biomass was enhanced at higher solution pH values. It is worth noting that the qm value at pH 4.0 was close to the amount of the biomass binding sites, 2.25 mmol/g, as determined from titrations [17]. The control samples demonstrated that the very high uranium uptake could not be attributed to micro-precipitation. 3.2. Desorption of uranium by HCl. The uranium-loaded biomass was eluted by various elutants, NaHCO3, (NH4)2SO4 and by mineral acids. It was established that the diluted mineral acids, such as H2SO4, HNO3 and HCl, were effective in uranium desorption and that no significant biomass damage resulted after several sorption-desorption cycles. The experimental results for the elution of the uranium-loaded biomass with various initial uranium loading by 0.1 N HCl are presented in Figure 3. 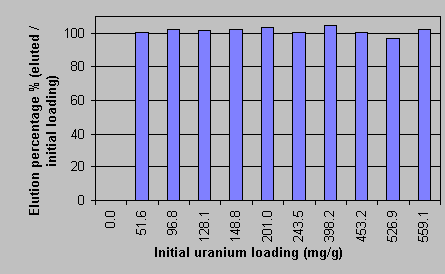 The Y axis in Figure 3 stands for the percentage of the eluted uranium metal over the initial uranium loading on the biomass. It could be noticed that the elution percentage values are close to unity within 4% error range, indicating that the elution with 0.1 N HCl was complete. The biomass weight loss during the acidic desorption process was less than 5%. The biomass was also protonated at the same time and it was thus ready for the next cycle of uranium biosorption uptake. 3.3. Uranium sorption and elution in a packed biomass column. In the biosorption of uranium in the column packed with Sagarsum biomass, approximately 10 L of 238 mg/L uranium solution was processed before the column breakthrough point occurred which was arbitrarily established at 1.0 mg/L of uranium in the column outlet. In this case, the column residence time was approximately 49 minutes and the total amount of 2,380 mg of uranium was accumulated on 22.64 g (dry) biomass. That gives the column overall uranium biosorption capacity of 105 mg U/g (dry biosorbent), including the only partially saturated portion of the dynamic sorption zone still inside at the column breakthrough (outlet 1 mgU/L). The results are illustrated in Figure 4 where the uranium concentration in the column outlet is plotted vs. the number of the column volumes that passed through the column. The column breakthrough took place at 36.5 bed volumes passing through. 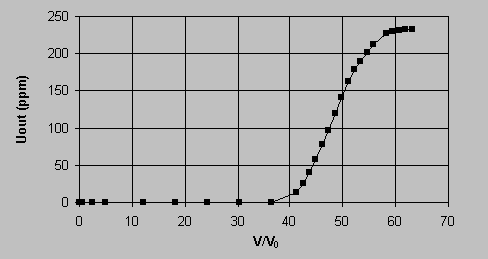 The elution curve for the column acid wash and recovery of uranium is shown in Figure 5. The uranium concentration in the elution acid (0.1N) at the outlet of the column was plotted against the volume of the elutant passing through the column. The narrow peak, about 6000 mg/L average concentration in 400 mL volume, and the low residual uranium concentration (< 1 ppm) indicated a highly efficient and complete column elution. After one month of continuous sorption-regeneration operation (5 cycles), no significant damage in biomass structures was observed. 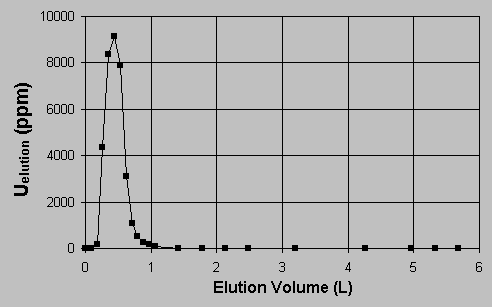 4. Discussion 4.1. The effect of pH on uranium biosorption mechanisms and the maximum sorption capacity. For algal biomass, ion exchange has been considered as a main mechanism responsible for metal sequestering [18, 19, 20]. The ion exchange mechanism for uranyl ion binding to the biomass is complicated by the fact that the uranium cation UO22+ is hydrolyzed in aqueous solutions within the pH range of the sorption system studied here. Partioning of the hydrolysed uranium species depends on the solution pH and on the total uranium concentration in the solution. In the range of acidic to near neutral pH values, four major hydrolysed complex ions, UO22+, (UO2)2(OH)22+, UO2OH+, (UO2)3(OH)5+ and a dissolved solid schoepite exist in the solution [21]. The hydrolysis equilibrium constants are pK = 5.8 for UO2OH+, pK = 5.62 for (UO2)2(OH)22+ and pK = 15.63 for (UO2)3(OH)5+[22]. The equilibrium coposition calculations could be carried out by the computer program MINEQL+ [21]. At pH 4.0, all hydrolyzed ions UO2OH+, (UO2)3(OH)5+ and (UO2)2(OH)22+ existed in the solution within all the present experimental concentration ranges of uranium. The percentage of the free ion UO22+ decreased while that of (UO2)2(OH)22+ increased with the increase in the total uranium concentration. The two monovalent ions took about 10 - 15 % of the total in all concentration ranges. According to Collins and Stotzky [23], the hydrolyzed species can apparently be sorbed better than the free hydrated ions. Particularly the monovalent ions, compared with the divalent hydrolyzed ions, have even higher affinity to the biomass in ion exchange with protons because they could replace single protons on separate binding sites in the biomass. The binding of the hydrolyzed ions onto the biomass would drive the hydrolysis equilibrium toward the formation of hydrolyzed complex ions when the hydrolyzed proton ions, H3O+s, were neutralized by the added LiOH to the system to maintain the constant solution pH 4.0. Eventually, the uranium would be sorbed on the binding sites in the form of hydrolyzed ions. When the hydrolyzed ions exchanged with protons, the ion exchange stoichiometry would be U/H+ = 1 : 1 for UO2OH+, U/H+ = 3 : 1 for (UO2)3(OH)5+ and U/H+ = 2 : 2 for (UO2)2(OH)22+, comparing with a U/H+ = 1 : 2 exchange ratio for the free ion UO22+. In another word, the hydrolyzed uranyl ions have a higher binding capacity on the biomass than the free ions. When the hydrolysed ions become prodominant in the ion exchange, the maximum molar uranium uptake could thus become close to or even higher than the value for the total binding capacity in the biomass, i.e. 2.25 meq/g. The extremely high experimental qm value of 2.4 mmol/g at pH 4.0 (Table 1) may be the demonstration of this case. With decreasing system pH, the percentage of UO22+ in the solution increased accordingly. The lower pH suppressed the enhancement of uranium biosorption occurring normally because of the hydrolyzed ions. When the pH became low enough, for example at pH 2.6, the divalent free UO22+ became the dominant ion form in the solution for a wide uranium concentration range from 0.3 to 1000 mg/L. Apart from a less preferable ion exchange ratio of U / H+ = 1 : 2, some binding sites which are far away from other sites may not be available for the divalent UO22+ which needs to exchange with two protons. This leads to an even lower uranium binding capacity at low pH. On the other hand, the non-ionic dissolved solid schoepite starts appearing in the solution when the pH is too high. The uranium sorption may be hindered by the decrease in ion concentration in this situation. For example, Guibal et al. [5] observed a decrease in uranium uptake by filamentous fungus biomass at pH 6.0. In summary, the biosorption of uranium on Sargassum biomass is a ion exchange process between the uranium ions and protons introduced to the biomass binding sites during the acid pre-treatment and biomass regeneration process. The hydrolysis of uranium ions, which is dependent on the solution pH, increased the uranium uptake by forming monovalent hydrolyzed complex ions. The number of available binding sites in the biomass for hydrolyzed ions was twice or more that for the divalent free UO22+ ions. Correspondingly, at pH 4.0, the maximum uranium uptake was as high as 566 mg/g or 2.38 mmol/g, which is quite close to the total amount of biomass binding sites. The present work demonstrates that uranium can be very effectively removed from the uranium-containing solution by the continuous-flow biosorption process. The uranium-laden Sargassum-based biosorbent can be conveniently eluted from the sorption column with a small volume of an HCl (0.1 N) wash which concentrates the metal. The high efficiency of biosorption and elution, low biomass damage and stability over a prolonged operation time make the new biosorption process an effective alternative for uranium pollution control which is coupled with the possibility of a feasible uranium metal recovery. 5. Glossary C0 Column feeding concentration (mmol/L) Ci, Cf Initial and final metal concentration (mmol/L) Cdes. Metal concentration in the eluant solution (mmol/L) F Flowrate passing through column (mL/hr) q Metal uptake (mmol/g) qdes. Eluted metal content per gram of biomass (mg/g) V Solution volume (L) Vb Column empty bed volume (mL); W Biomass weight (g) References [1] White, S.K. J. Am. Water Works Assoc. 1983, 75, 374. [2] Laul, J.C. Radioanal. Nucl. Chem. Articles 1992, 156, 235. [3] Benedict, B.; Pigford, T.H.; Levi, H.W. Nuclear Chemical Engineering,
McGraw-Hill
[4] Volesky, B.; Tsezos, M. 1981, US Patent 4 320 093; Canadian
Patent 1 143 007
[5] Guibal, E.; Roulph, C.; Le Cloirec, P. Water Res. 1992, 26, 1139-45. [6] Macaskie, L.E.; Empson, R.M.; Cheetham, A.K.; Grey, C.P.; Skarnulis,
A.J. Science
[7] Munroe, N.D.H.; Bonner, J.D.; Williams, R.; Pattison, K.F.; Norman,
J.M.; Faison, B.D.
[8] Hu, M.Z.-C.; Norman, J.M.; Faison, N.B.; Reeves, M. Biotechnol.
Bioeng. 1996,
[9] Horikoshi, T.; Nakajima, A.; Sakaguchi, T. Agric. Biol. Chem. 1979, 332, 617. [10] Byerley, J.J.; Scharer, J.M.; Charles, A.M. Chem. Eng. Journal 1987, 36, B49-B59. [11] Kuyucak, N.; Volesky, B. In Biosorption of Heavy Metals
; Volesky, B., ed.;
[12] Volesky, B.; Holan, Z.R. Biotechnol. Prog. 1995, 11, 235-250. [13] Kuyucak, N.; Volesky, B. Biorecovery 1989, 1, 189-204. [14] Leusch, A.; Holan, Z.R.; Volesky, B. J. Chem. Tech. Biotechnol. 1995, 62, 279-288. [15] Aldor, I.; Fourest, E.; Volesky, B. Can. J. Chem. Eng. 1995, 73, 516-522. [16] Edgington, D.N.; Gorden, S.A.; Thommes, M.M.; Almodovar, L.R. Limnol.
Ocean.
[17] Fourest, E.; Volesky, B. Environ. Sci. Technol. 1996, 30, 277-282. [18] Crist, R.H.; Oberholser, K.; Schwartz, D.; Marzoff, J.; Ryder,
D.; Crist, D.R. Environ.
[19] Crist, D.R.; Crist, R.H.; Martin, J.R.; Watson, J. In Metals-Microorganisms
[20] Schiewer, S.; Volesky, B. Environ. Sci. Technol. 1995, 29, 3049-3058. [21] Schecher, W.D. MINEQL+ : A Chemical Equilibrium Program for
Personal
[22] Baes, C.F.J.; Mesmer, R.E. The Hydrolysis of Cations, Wiley-Interscience,
John
[23] Collins, Y.E.; Stotzky, G. Appl. Environ. Microbiol. 1992,
58, 1592-1600.
[ Bact to the Beginning ] [ Bact to the Index ] [Biosorption Home] [ Dr. Volesky's Home ] |
||||||||||||||||||||
|
By Jinbai Yang (E-mail: cyya@musica.mcgill.ca ) |